Autophagy – the housekeeper in every cell that fights aging
By James P Watson and Vince Giuliano
Background and introduction
There is a wide variety of genetic manipulations, pharmacologic manipulations, and nutrient manipulations that have been shown to alter lifespan in model organisms. These include caloric restriction, “loss of function” mutations, “gene knock out” models, phytochemicals, and drugs that down regulate aging pathways (mTOR, insulin/IGF-1, etc.). It also includes “gain of function mutations”, transgenic models, phytochemicals, and drugs that up regulate longevity promoting pathways (AMPK, FOXO, Klotho, etc.). At first glance, all these interventions may seem to be unrelated, suggesting that aging is a multifactorial problem with no common denominator to longevity. On further examination, however, there is a common denominator to all of these interventions – autophagy. Autophagy (“self eating”) is an old, evolutionarily conserved stress response that is present in all living cells. Like apoptosis, autophagy is a programmed response and has several sub-pathways. Unlike apoptosis, autophagy promotes life rather than death. Recent discoveries have shown that almost every genetic, dietary, and pharmacologic manipulation proven to extend lifespan activates autophagy as part of its mechanism of action.
Autophagy is the way your cells “clean house” and “recycle the trash”. Along with the ubiquitin proteasome system, autophagy is one of the main methods that cells use to clear dysfunctional or misfolded proteins. Autophagy can clear any kind of trash: intracellular viruses, bacteria, damaged proteins, protein aggregates and subcellular organelles. Although autophagy has long been known to exist, only recently has there been a clear understanding of the genes and pathways related to it. This recent evidence suggests that the declining efficacy of autophagy may be a driver of many of the phenotypic phenomena of aging. This blog entry explores the “evidence for the autophagy theory of aging” and builds a strong case that defective autophagy is a central driver for age-related diseases and aging itself.
Autophagy now appears to be a downstream event following insulin/IGF-1 pathway down-regulation, mTOR inhibition, Klotho activation, AMPK activation, Sirtuin dependent protein deacetylation, and histone acetyl transferase inhibition. Autophagy explains in part, the beneficial effects of caloric restriction, caffeine, green tea, rapamycin, resveratrol, metformin, spermidine, lithium, exercise, hypoxia, Torin-1, trehalose, and a host of other natural and synthetic compounds.
There is much stronger evidence of a link between autophagy activation and longevity than there is with any other longevity interventions such as exogenous anti-oxidant supplementation, endogenous anti-oxidant up regulation, micronutrient replacement, hormone replacement, anti-inflammatory therapy, telomerase activation, or stem cell therapy. For this reason, we have listed below the top reasons why “eating yourself for dinner” mauy well be the best way to promote health and longevity.
What is autophagy?
Biological entities employ various mechanisms to keep themselves functioning healthily, including mechanisms to get rid of defective or no longer wanted components. Inter and intra-cell signaling can drive a cell to destroy itself, for example (cell apoptosis). Short of apoptosis, on the cell level there are several mechanisms for getting rid of defective or no longer needed components including organelles and proteins. From the 2008 publication Autophagy and aging: “All cells rely on surveillance mechanisms, chaperones and proteolytic systems to control the quality of their proteins and organelles and to guarantee that any malfunctioning or damaged intracellular components are repaired or eliminated [1,2]. Molecular chaperones interact with unfolded or misfolded proteins and assist in their folding [3]. However, if the extent of protein damage is too great, or the cellular conditions are not adequate for re-folding, the same molecular chaperones often deliver proteins for degradation. Two proteolytic systems contribute to cellular clearance: the ubiquitin-proteasome and the lysosomal systems [4].” Autophagy is concerned with the lysosomal system and involves the “degradation of any type of intracellular components including protein, organelles or any type of particulate structures (e.g. protein aggregates, cellular inclusions, etc.) in lysosomes(ref)”
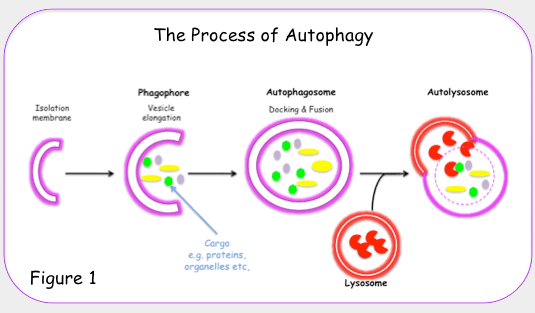
“Autophagy, or autophagocytosis, is a catabolic process involving the degradation of a cell’s own components through the lysosomal machinery. It is a tightly regulated process that plays a normal part in cell growth, development, and homeostasis, helping to maintain a balance between the synthesis, degradation, and subsequent recycling of cellular products. It is a major mechanism by which a starving cell reallocates nutrients from unnecessary processes to more-essential processes. Autophagy is an evolutionarily conserved mechanism of cellular self-digestion in which proteins and organelles are degraded through delivery to lysosomes. Defects in this process are implicated in numerous human diseases including cancer(ref).”
Top 16 Key Facts about Autophagy
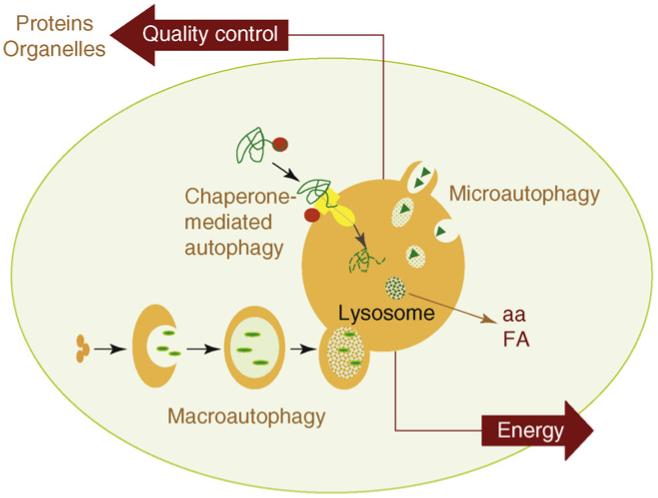
There are three main pathways of Autophagy – Macroautophagy, Microautophagy, and Chaperone-mediated Autophagy (CMA).
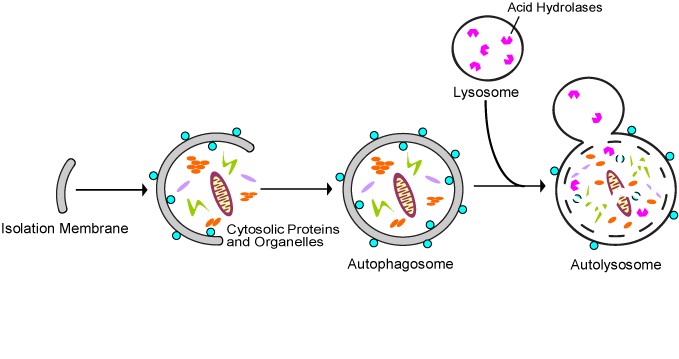
All 3 autophagy pathways are constitutively active (i.e. they can occur at basal levels) but can also be up regulated by cellular stress). Macroautophagy is the primary “broom” that sweeps the house. Macroautophagy is initiated when the material to be removed is tagged with ubiquitin. This signals a complex series of molecular events that leads to the formation of a membrane around the material to be removed and recycled. This membrane formation around the debris is called a autophagosome. Once formed, the autophagocome fuses with a lysosome to form an autolysosome. Once fusion occurs, the acid hydrolases found inside the lysosomes start digesting the damaged proteins and organelles. When damaged mitochondria are digested by macroautophagy, it is called mitophagy, which is a specific type of macroautophagy. Macro-autophagy can also remove and recycle mutated or free-radical damaged proteins or protein aggregates. Macroautophagy and other sub cellular organelles (peroxisomes, endoplasmic reticulum, etc.) Even part of the cell nucleus can undergo autophagy (called “piecemeal microautophagy of the nucleus” – PMN).
Chaperone-mediated autophagy (CMA) is a specific mechanism of autophagy that requires protein unfolding by chaperones. The other two mechanisms do not require protein unfolding (macroautophagy and microautophagy). Since protein aggregates cannot be unfolded by chaperone proteins, both the ubiquitin-proteasome system and chaperone-mediated autophagy are unable to clear these protein aggregates. For this reason, macroautophagy may be the most important pathway for preventing Alzheimer’s disease, Parkinson’s disease, Fronto-temporal dementia, and all of the other neurodegenerative diseases associated with protein aggregate accumulation.
Microautophagy is essentially just an invagination (folding in) of the lysosomal membrane and does not require the formation of an double-membrane autophagosome. Both CMA and microautophagy appear to play a minor role in “house keeping”. Here are diagrams of these types of autophagy.
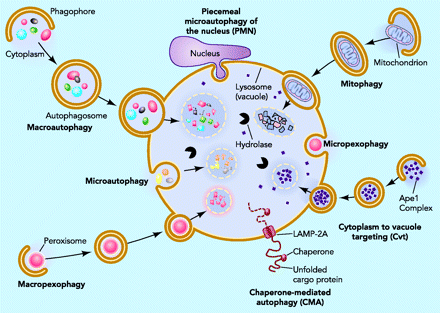
{kind=link}
2. Autophagy is the only way to Get Rid of Old Engines – i.e. damaged mitochondria
Autophagy is the best way to get rid of bad mitochondria without killing the cell. The process is called “mitophagy.” Since bad mitochondria produce most of the “supra-hormetic doses of ROS”, this is really, really, important. This is explained in our recent blog entries related to mitochondria, Part 1, and Part 2. For brain cells, heart cells, and other post mitotic cells that we all want to “hang on to”, mitophagy is probably the most important anti-aging value of mitophagy. Bad mitochondria are phosphorylated by the kinase PINK1. Then these bad mitochondria are ubiquinated by the E3 ligase Parkin. The ubiquinated bad mitochondria are then selectively destroyed by mitophagy, which is a form of macroautophagy.
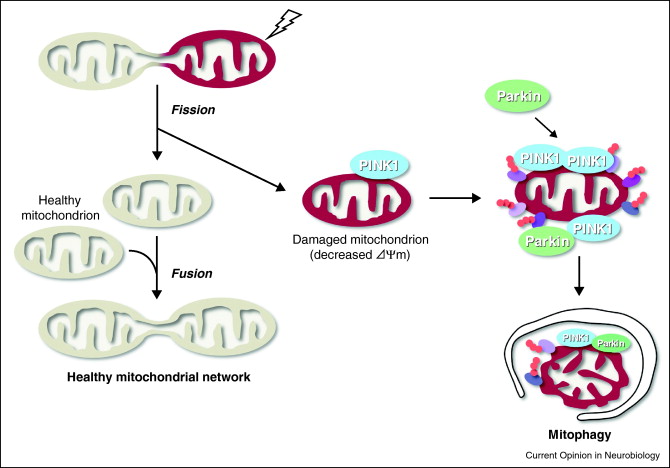 Mitophagy Image source
Mitophagy Image source
{kind=link}
The 2007 publication Selective degradation of mitochondria by mitophagy reviews the topic. “Mitochondria are the essential site of aerobic energy production in eukaryotic cells. Reactive oxygen species (ROS) are an inevitable by-product of mitochondrial metabolism and can cause mitochondrial DNA mutations and dysfunction. Mitochondrial damage can also be the consequence of disease processes. Therefore, maintaining a healthy population of mitochondria is essential to the well-being of cells. Autophagic delivery to lysosomes is the major degradative pathway in mitochondrial turnover, and we use the term mitophagy to refer to mitochondrial degradation by autophagy. Although long assumed to be a random process, increasing evidence indicates that mitophagy is a selective process.”
3. Autophagy is the best Way to Get Rid of Junk. – protein aggregates, etc.
Autophagy is the best way to get rid of protein aggregates like those associated with all of the neurodegenerative diseases, like amyloid beta, tau tangles, alpha synuclein aggregates, TDP-43 aggregates, SOD aggregates, and Huntington protein aggregates. These aggregates are NOT digested via the ubiquitin-proteasome system, since they cannot be “unfolded”. For this reason, autophagy is probably the most important cellular mechanism for clearing protein aggregates found in neurodegenerative diseases. Autophagy can also clear out bad cytoplasm (Cvt), endoplasmic reticulum, peroxisomes (micro and macropexophagy), Golgi apparatus, and even damaged parts of the nucleus (PMN). See for example (2012) Degradation of tau protein by autophagy and proteasomal pathways and (2009) Autophagy protects neuron from Abeta-induced cytotoxicity
Autophagy is protective by quietly getting rid of multiple other unwanted substances. For example, it protects against alcohol-induced liver damage. Consider what is going on in this diagram from the 2011 publication The emerging role of autophagy in alcoholic liver disease:
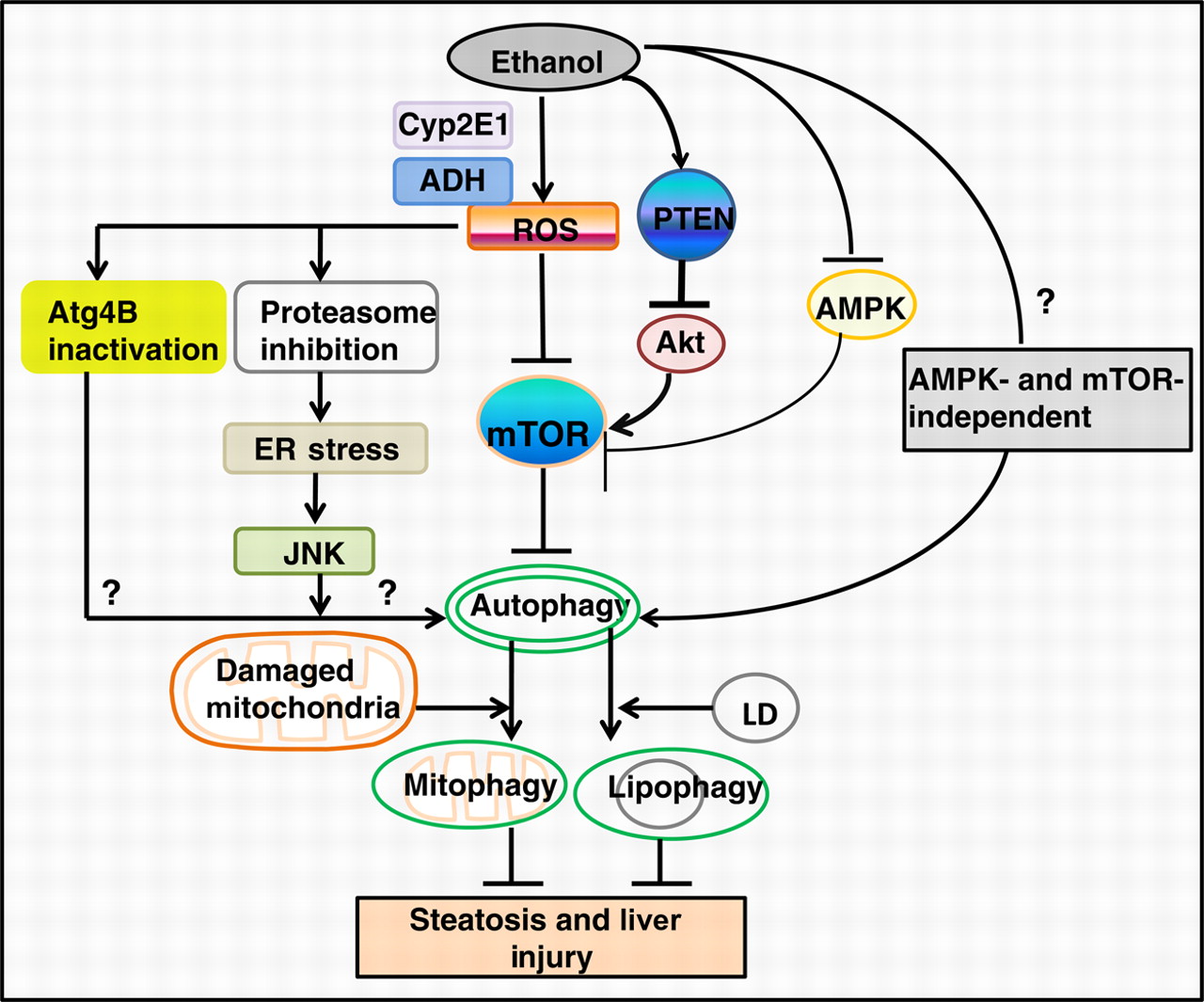 Image source “Alcohol consumption causes hepatic metabolic changes, oxidative stress, accumulation of lipid droplets and damaged mitochondria; all of these can be regulated by autophagy. This review summarizes the recent findings about the role and mechanisms of autophagy in alcoholic liver disease (ALD), and the possible intervention for treating ALD by modulating autophagy(ref).”
Image source “Alcohol consumption causes hepatic metabolic changes, oxidative stress, accumulation of lipid droplets and damaged mitochondria; all of these can be regulated by autophagy. This review summarizes the recent findings about the role and mechanisms of autophagy in alcoholic liver disease (ALD), and the possible intervention for treating ALD by modulating autophagy(ref).”
{kind=link}
4. Aging = Autophagy decline.
According to the 2008 publication Autophagy in aging and in neurodegenerative disorders: “Growing evidence has indicated that diminished autophagic activity may play a pivotal role in the aging process. Cellular aging is characterized by a progressive accumulation of non-functional cellular components owing to oxidative damage and a decline in turnover rate and housekeeping mechanisms. Lysosomes are key organelles in the aging process due to their involvement in both macroautophagy and other housekeeping mechanisms. Autophagosomes themselves have limited degrading capacity and rely on fusion with lysosomes. Accumulation of defective mitochondria also appears to be critical in the progression of aging. Inefficient removal of nonfunctional mitochondria by lysosomes constitutes a major issue in the aging process. Autophagy has been associated with a growing number of pathological conditions, including cancer, myopathies, and neurodegenerative disorders.”
The relationship of autophagy decline to hallmarks of aging has been known for a long time and have been best studied in liver cells. The auto florescent protein lipofuscin is the oldest and simplest biomarker of declining autophagy and represents undigested material inside of cells. The Lewy bodies seen in several neurodegenerative diseases (including “Parkinson’s disease with dementia”) are also biomarkers of declining autophagy and may specifically be due to “declining mitophagy”. Declining autophagy is particularly important in post-mitotic cells such as those in the brain, heart, and skeletal muscle where very little cell regeneration via stem cells occurs. For mitotic tissues such as the GI tract, bone marrow, and skin, autophagy decline may not be as detrimental, since apoptosis is another normal method for getting rid of bad cells.
The failure of autophagy with aging has several possible causes:
a. Fusion problems – Autophagic vacuoles accumulate with age in the liver. This may be due to a problem of fusion between the autophagosomes and the lysosomes.
b. Glucagon deficiency – Glucagon is a hormone that enhances macroautophagy. “—the stimulatory effect of glucagon [on autophagy] is no longer observed in old animals. See item (b) in the next list below.(ref)“
c. Negative signaling via the Insulin receptor – Insulin activates the Insulin/IGF-1 pathway which activates mTOR. mTOR activation inhibits autophagy (see below). Even in the absence of insulin, there is up-regulation with aging of the insulin/IGF-1 signaling via the insulin receptor tyrosine kinase. This would activate mTOR.
d. Inadequate turnover of damaged mitochondria – Mitophagy decline may be one of the mechanisms that is responsible for the decline in autophagy with aging. Specifically, if mitophagy does not keep up with the demand for damaged mitochondrial clearance, a higher baseline ROS would occur, which would damage proteins, cell membrane lipids, and cell nucleus DNA.
e. Energy compromise – With aging, there is a decline in energy production by the cells. This may be one of the reasons for the decline in autophagy seen in aging.
Here is a depiction of some of the main problems associated with decline of autophagy in aging:
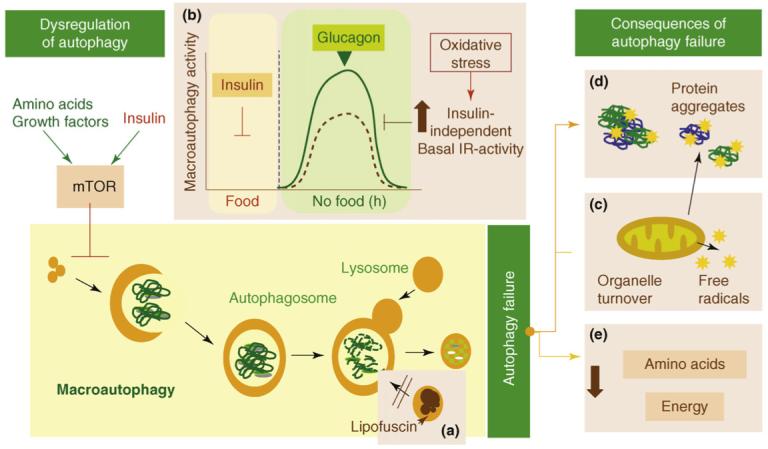
Some consequences of failure of autophagy with aging “Possible causes and consequences of the failure of macroautophagy in old organisms are depicted in this schematic model (brown boxes” Image source
(a) The accumulation of autophagic vacuoles with age could result from the inability of
lipofuscin- loaded lysosomes to fuse with autophagic vacuoles and degrade the sequestered content.
(b) In addition, the formation of autophagosomes in old cells might be reduced because of the inability of macroautophagy enhancers (such as glucagon) to induce full activation of this pathway. The stimulatory effect of glucagon is compromised in old cells because of maintained negative signaling through the insulin receptor (IR) even under basal conditions (i.e. in the absence of insulin). Maintained insulin signaling would activate mTOR, a known repressor of macroautophagy.
(c) Inadequate turnover of organelles, such as mitochondria, in aging cells could increase levels of free radicals that generate protein damage and
(d) Aging could also potentiate the inhibitory signaling through the insulin receptor.
(e) An age-dependent decline in macroautophagy can also result in energetic compromise of the aging cells.
5. Genetic manipulations that increase lifespan in all model organisms stimulate autophagy.
Knocking out macroautophagy takes away at least 50% of the long-lived mutant’s added lifespan. This same “loss of longevity” is seen with Caloric restriction in “macroautophagy knockouts”. The following diagram shows how important autophagy is in long-lived mutant nematodes and how this is important for increasing lifespan, reducing cellular damage, and increasing function.
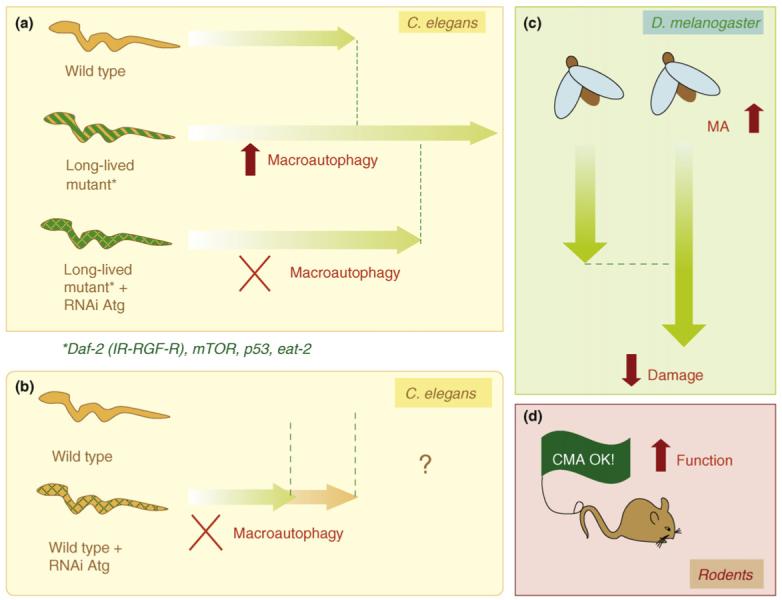
The most well studied “mutants” are model organisms where one of the following pathways are altered by a gene mutation or a gene knock out. When an additional “knocking out” of an autophagy gene is done, approximately 1/2 of the added lifespan of the long lived mutants (vs wild type) appears to be “wiped out” by loosing autophagy. Similar findings occur in “macroautophagy knock-outs” subjected to caloric restriction, etc. This suggests to me that 1/2 of the benefits of caloric restriction are due to stimulating autophagy. Caloric restriction down regulates all of the”nutrient sensing pathways that are negative regulators of autophagy” and up regulates other “ nutrient sensing pathways that are positive regulators of autophagy”. The following interconnected “nutrient -sensing pathways” affect macroautophagy:
a. IGF-1: two mechanisms:
i. decreasing Insulin-IGF-1 pathway => tyrosine kinase => inhibits Akt phosphorylation of TSC => inhibition of raptor in mTOR complex
ii. decreasing insulin/IGF-1 pathway => Foxo transcription factor translocation to nucleus => FOXO stimulates autophagy via activating two autophagy genes – LC3 and BNIP3.
b. mTOR: three mechanisms account for the activation of autophagy by mTOR inhibition
i. mTOR inhibition => decreases phosphorylation of Atg1 (aka ULK1/2). Also decreases phosphorylation of Atg13 and Atg17. Phosphorylation of ULK1/2, Atg13, and Atg17 inhibits autophagy initiation.
ii. decreasing mTOR pathway => decreases phosphorylation of 4EBP1 => blocks effect of eIF4F => autophagy activation.
iii. decreasing mTOR pathway => decreases phosphorylation of S6K => S6K no longer active => inhibition of autophagy.
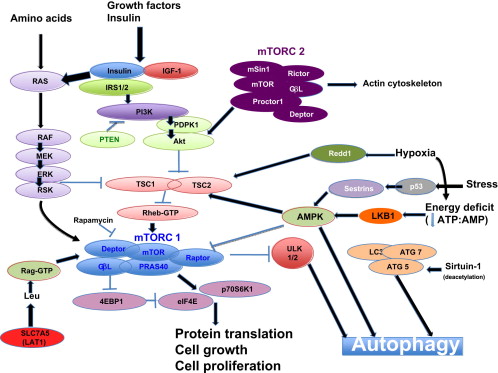
Signaling pathways that affect autophagy Image source
“The (mammalian) target of rapamycin (mTOR) is a primordial negative regulator of autophagy inorganisms from yeast to man. mTOR is inhibited under starvation conditions, and this contributes to starvation-induced autophagy via activation of mTOR targets Atg13, ULK1, and ULK2. This inhibition can be mimicked by mTOR inhibitory drugs like rapamycin (Ravikumar et al., 2010). One of the important pathways regulating mTOR is initiated when growth factors like insulin-like growth factor bind to insulin-like growth factor receptors (IGF1R) (Figure 2). These receptors signal, via their tyrosine kinase activities, to effectors like the insulin receptor substrates (IRS1 and IRS2), which in turn activate Akt. Akt inhibits the activity of the TSC1/TSC2 (proteins mutated in tuberous sclerosis) complex, a negative regulator of mTOR. In this way, IGF1R signaling activates mTOR and inhibits autophagy, and the converse occurs when nutrients are depleted(ref).”
c. Ras/PKA: decreasing Protein Kinase A pathway (aka Ras/cAMP) => decreases phosphorylation of 3 autophagy proteins (Atg1, Atg13, Atg18).
d. PKB/Akt: decreasing Protein Kinase B pathway (aka PkB/Akt or Sch9) => reduces inhibition of TSC-1 => decreased mTOR activity.
e. Sirtuin 1: CR activates Sirtuin 1 => deacetylation of several autophagy gene products: Atg5, Atg7, Atg8/LC3. Sirt1 also activates AMPK, activates FOXO3a, and inhibits mTOR via TSC-1/2
f. AMPK: AMPK pathway (aka LKB1-AMPK) activates autophagy via two methods:
i. AMPK activation => phosphorylates TSC2 and raptor => inhibits TORC1 (this requires glucose starvation).
ii. AMPK activation => direct phosphorylation of Atg1 (aka ULK1) => autophagy activation (this does NOT require glucose starvation).
g. Less-important pathways:
i. Rim15: increasing Rim15 Kinase pathway => Msn2 and Msn4 transcription factor translocation to nucleus => inhibits mTOR, PKA, and PKB pathways.
ii ERK1/2: ERK pathway – the extracellular signal-regulated kinase (ERK) also mediates starvation-induced autophagy. (see #6 below for more details)
iii. JNK: JNK pathway – This is a MAPK that mediates starvation-induced autophagy. (see #6 below for more details).
The main pathways are depicted in the following diagram of how Calorie Restriction works (Ras/PKA and less important pathways not depicted).
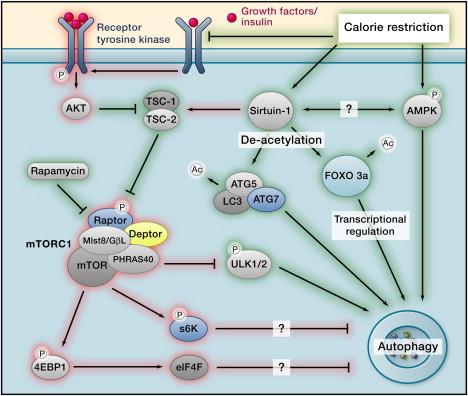
Autophagy regulation Image source
6. There are many other pathways that regulate autophagy that are not dependent on “nutrient sensing pathways.”
(i.e. not those described above).
Although caloric restriction or fasting are clearly the most “potent” autophagy stimulators, since they can activate macroautophagy via the above “nutrient sensing pathways“, there are many other pathways that can activate autophagy. Here an explanation of the roles of the key kianses involved:
a. PI3Ks and Akt – PI3Ks are kinases that are mainly activated by growth factors, not starvation. There are 3 classes of PI3Ks and the Class III PI3Ks directly positively activate autophagy (Vps34) whereas the Class I PI3Ks indirectly inhibit autophagy via mTOR and Akt.
b. MAPKs – Mitogen-Activated Protein Kinase – these are kinases that are mainly activated by growth factors, not starvation. There are 3 classes:
i. ERK – Extracellular signal-Regulated Kinases (ERK) positively regulate autophagy by maturing autophagic vacuoles. EKR also seems to specifically be involved with mitochondrial-specific autophagy (i.e. mitophagy). Mitochondrial ERK may help protect from neurodegenerative diseases. Cancer cells also activate mitochondrial ERK to cause chemoresistance. ERK is activated downstream from Ras. Ras activates Raf, which activates MEK. MEK phosphorylates and activates ERK1 and ERK2.
This is the mechanism by which you can kill cancer with soy extracts, capsaicin, and Cadmium. Here is how this works:
- Soyasaponins (found in soybeans) => activates ERK => autophagy-induced death in colon cancer cells
- Capsaicin (found in chili peppers) => activates ERK => autophagy-induced death in breast cancer cells
- Cadmium (toxic metal) => activates ERK => autophagy-induced death in mesangial cells
ii. p38 – p38 is a MAPK that is a tumor suppressor. p38 regulates autophagy but there is still controversy if it activates or inhibits autophagy.
iii. JNK – JNK is a MAPK that is activated by heat shock, osmotic shock, UV light, cytokines, starvation, T-cell receptor activation, neuronal excitotoxic stimulation, and ER stress. With starvation, JNK does not phosphorylate Bcl-2, which prevents it from binding to beclin 1. Beclin 1 can then induce autophagy. Bcl-2 is an anti-apoptotic protein and can prevent apoptosis. There are multiple phosphorylation sites on Bcl-2. The degree by which JNK phosphorylates/dephosphorylates Bcl-2 may determine cell fate – i.e. apoptosis (death) vs autophagy (survival). See (2011) The Beclin 1 network regulates autophagy and apoptosis.
c. PKC – Protein Kinase C (PKC) is a family of kinases that were once thought to be associated mostly with apoptosis/anti-apototis. Recent research has shown that PKCs also play a role in autophagy. The effects of PKC depend on if the cellular stress is acute or chronic. For instance, PKCg is an example of one of the PKCs where it stimulates autophagy with acute, short periods of hypoxia (via JNK activation) but suppresses autophagy with chronic hypoxia (via Caspace-3). Another PKC, PKC0 is involved with ER-stress induced autophagy. Acadesine (AICAR) induces autophagy via a PKC/Raf1/JNK pathway. Acadesine (AICAR) in combination with GW1516 has shown to improve endurance-type exercise by converting fast-twitch muscle fibers into the more energy-efficient, fat-burning, slow-twitch muscle fibers. These two compounds turned on 40% of the genes that were turned on when exercise + GW1516 were used together. For this reason, acadesine (AICAR) has been termed an “exercise mimetic” and has been banned for use by athletes, since it is a performance enhancing drug, even though it is very safe. The mechanism of action of AICAR may be in part its induction of autophagy.
d. Endoplasmic Reticulum Stress Kinases (i.e. the ER unfolded protein response) – Several kinases involved with the endoplasmic reticulum unfolded protein response (ER-UPR) have been found to activate autophagy. They include the following:
i. IRE-1 – Inositol-requiring enzyme (IRE1) is one of the first proteins activated by the ER-UPR. It up regulates autophagy genes (Atg5, 7, 8, 19).
ii. PERK – PERK must phosphorylate the eukaryotic initiation factor 2alpha (eIF2alpha) for LC3 conversion with ER-UPR induced autophagy. PERK also up regulates Atg5.
iii. CaMKKbeta – ER stress results in calcium release from the ER. This Ca++ release induces autophagy via the Ca dependent kinases. The main one is called Ca/Calmodulin-dependent kinase beta (CaMKKbeta). This is an “upstream activator” of AMPK, which in turn inhibits mTOR. This is how calcium can induce autophagy.
iv. DAPK1 – Death-associated protein kinase 1 (DAPK1) is another Ca++/Calmodulin-regulated kinase that is important in ER-UPR induced autophagy. It induces autophagy by phosphorylating beclin 1, which is necessary for autophagosome formation.
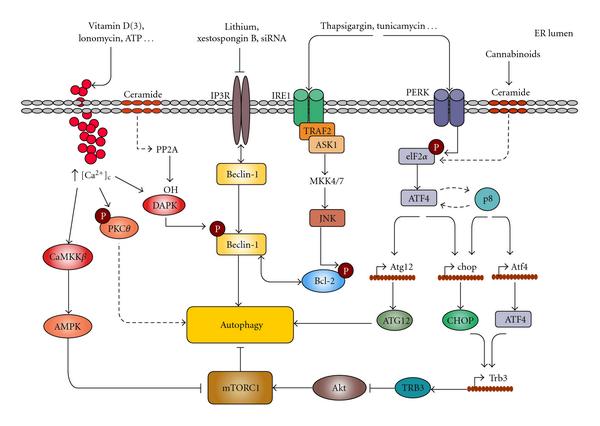
Mechanisms connecting ER stress and autophagyImage Source “Mechanisms connecting ER stress and autophagy. Different ER stresses lead to autophagy activation. Ca2+ release from the ER can stimulate different kinases that regulate autophagy. CaCMKK phosphorylates and activates AMPK which leads to mTORC1 inhibition; DAPK phosphorylates Beclin-1 promoting its dissociation from Bcl-2; PKCθ activation may also promote autophagy independently of mTORC1. Inositol 1,4,5-trisphosphate receptor (IP3R) interacts with Beclin-1. Pharmacological inhibition of IP3R may lead to autophagy in a -independent manner by stimulating its dissociation from Beclin-1. The IRE1 arm of ER stress leads to JNK activation and increased phosphorylation of Bcl-2 which promotes its dissociation from Beclin-1. Increased phosphorylation of eIF2 in response to different ER stress stimuli can lead to autophagy through ATF4-dependent increased expression of Atg12. Alternatively, ATF4 and the stress-regulated protein p8 promote the up-regulation of the pseudokinase TRB3 which leads to inhibition of the Akt/mTORC1 axis to stimulate autophagy(ref).”
7. Excess baseline ROS from bad mitochondria induces Mitophagy.
– ROS induces autophagy via a non-canonical pathway.
This may be the mitochondrial signal for “selective destruction” of damaged mitochondria. Exogenous ROS can also induce autophagy, however. For instance, there is evidence that abnormal levels of H202 in the cytoplasm will induce macroautophagy. Hydrogen peroxide induces a “non-canonical autophagy” that is “beclin-1 independent” but requires the JNK-mediated activation of Atg7. on of Atg7.
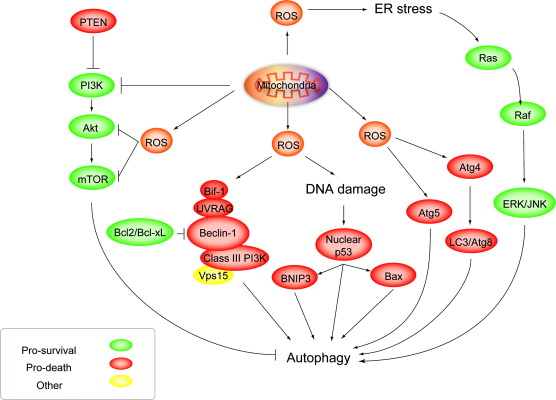
ROS induces autophagy: Roles of Akt, ERK, JNK and BeclinsImage source
8. Most all of the Pharmacologic manipulations that extend lifespan increase autophagy.
Here are some of the main ones:
a. Rapamycin – Autophagy explains most of the longevity and health benefits (mechanism of action) of Rapamycin
Since the protein kinase mTOR phosphorylates the 3 key autophagy initiating proteins (Atg1, Atg13, and Atg17), it is considered the “Master of Autophagy”. Rapamycin inhibits both TORC1 and TORC2. TORC1 inhibition is the the “direct” and primary mechanism by which rapamycin activates autophagy, but TORC2 inhibition has an “indirect” and independent method of activating autophagy via inhibiting Akt or Protein Kinase C. (This is why Blagonosky in NY likes rapamycin over TORC1-specific mTOR inhibitors).
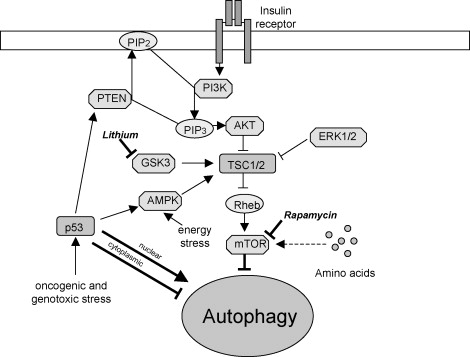
Image source mTOR and autophagy, showing impacts of lithium and rapamycin
b. Metformin – .Autophagy may explain as much as 50% of the benefits (mechanism of action) of Metformin.
Metformin activates AMPK and therefore stimulates autophagy via TORC1-dependent and TORC-1 independent methods (see above). For this reason, metformin is a good “autophagy drug”. Metformin probably has many other mechanisms of action, however, which cannot be explained by the induction of autophagy.
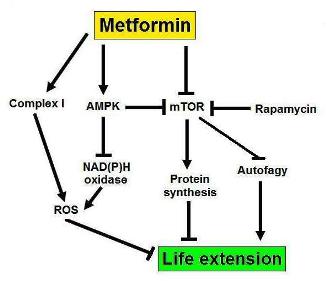
{kind=link}
c. Resveratrol – Resveratrol directly or indirectly activates the NAD+-dependent deacetylase, SIRT1.
SIRT1 activates autophagy by several different mechanisms, the 4 major ones being deacetylation of multiple cytoplasmic proteins including several involved with autophagy, such as ATG5, ATG7, and ATG8/LC3. SIRT1 also deacetylates the FOXO transcription factors (FOXO3a, FOXO, and FOXO4), but the FOXO proteins are not required for autophagy induction. It is likely that the effects of SIRT1 on FOXO deacetylation mediate other beneficial effects of resveratrol (not autophagy).
d. Spermidine – The benefits of spermidine can be partially explained by its effects on autophagy. Spermidine is a histone acetylase inhibitor. By inhibiting histone acetylase, spermidine allows for the up regulation of autophagy (Atg) genes. It appears that like resveratrol, spermidine also stimulates overlapping deacetylation reactions of cytoplasmic proteins. See the 2009 publication Autophagy mediates pharmacological lifespan extension by spermidine and resveratrol.
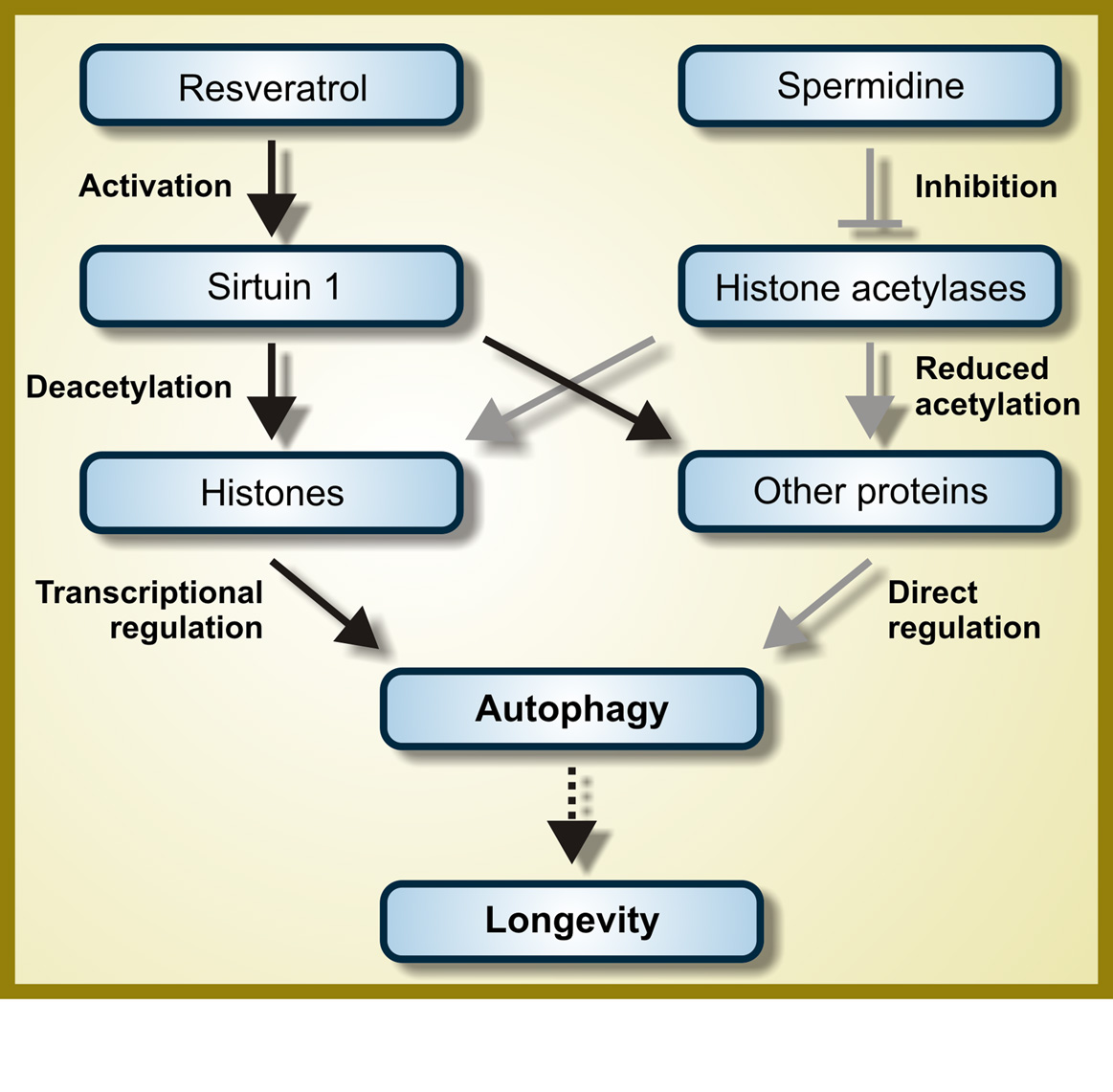
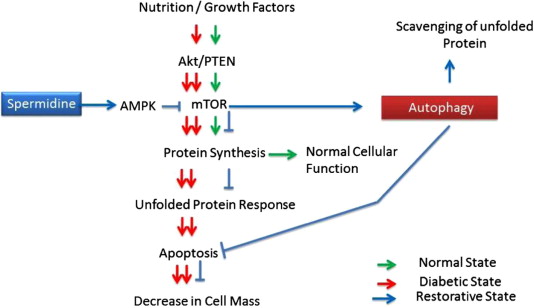
Spermidine and autophagy in normal and diabetic states Image source
e. Lithium – The beneficial effects of Lithium for aging and for bipolar illness may be mediated in part by autophagy(ref).
9. Exercise can both activate and inhibit autophagy.
For this reason, the benefits of exercise are mostly due to non-autophagy factors.
Decreased autophagy mechanisms with exercise: Exercise up regulates mTOR, especially resistance exercises like weight lifting. Exercise also activates the IGF-1 pathway by increasing growth hormone secretion by the pituitary gland, which then in turn stimulates IGF-1 production by the liver. IGF-1 inhibits autophagy via the Insulin/IGF-1/PI3K/Akt pathway.
Increased autophagy mechanisms with exercise: ROS increases with exercise. Since ROS activates autophagy, this is one mechanism by which exercise could activate autophagy, but it is unclear if this activates “selective mitochondrial destruction” this way (i.e. mitophagy).
Hypoxia also activates autophagy via a HIF-1a pathway. This would occur with exercise if you reached your anaerobic threshold during exercise or did IHT exercise (intermittent hypoxia with exercise).
Conclusion: Exercise can both inhibit and activate autophagy. This may be why it is difficult to show exactly how exercise prolongs lifespan.
10. Autophagy exercises anti-aging effects on postmitotic cells.
– There are primarily 5 cytoprotective effects:
- Reduced accumulation of toxic protein aggregates, described above
- Destroying bad mitochondria via mitophagy, described above
- Reduced apoptosis
- Reduced necrosis
- Improved hormesis
Cells that do not divide are particularly vulnerable to the build-up of protein aggregates seen in neurodegenerative diseases. Autophagy inducers such as rapamycin, rapalogs, valproate, and lithium have been shown to help in experimental models of Huntington’s disease, tauopathies, Alzheimer’s disease, and Parkinson’s disease.
When mitochondria are defective due to ROS-induced damage, asymmetric fission occurs, allowing for a good mitochondria and a bad mitochondria to “split up”. The bad mitochondria has a low membrane potential and is tagged by PINK1 and then ubiquinated by Parkin. At this point, it is recognized by the autophagy system and is destroyed by macroautophagy.
Autophagy also has an anti-apoptotic function in post mitotic cells. Autophagy helps damaged cells recover and thereby avoid apoptosis. Autophagy also has an “anti-necrosis” function in post mitotic cells.
Autophagy is also a stress response involving hormesis. Hormesis is how low (sublethal) doses of cellular stressors result in an up regulation of cellular stress adaptation mechanisms. See the blog entries Multifactorial hormesis II – Powerpoint presentation and Multifactorial Hormesis – the theory and practice of maintaining health and longevity. Autophagy has a hormetic dose response curve. Depending on the strength or duration of the stressor, autophagy or a negative consequence could ensue, as exemplified in this diagram:
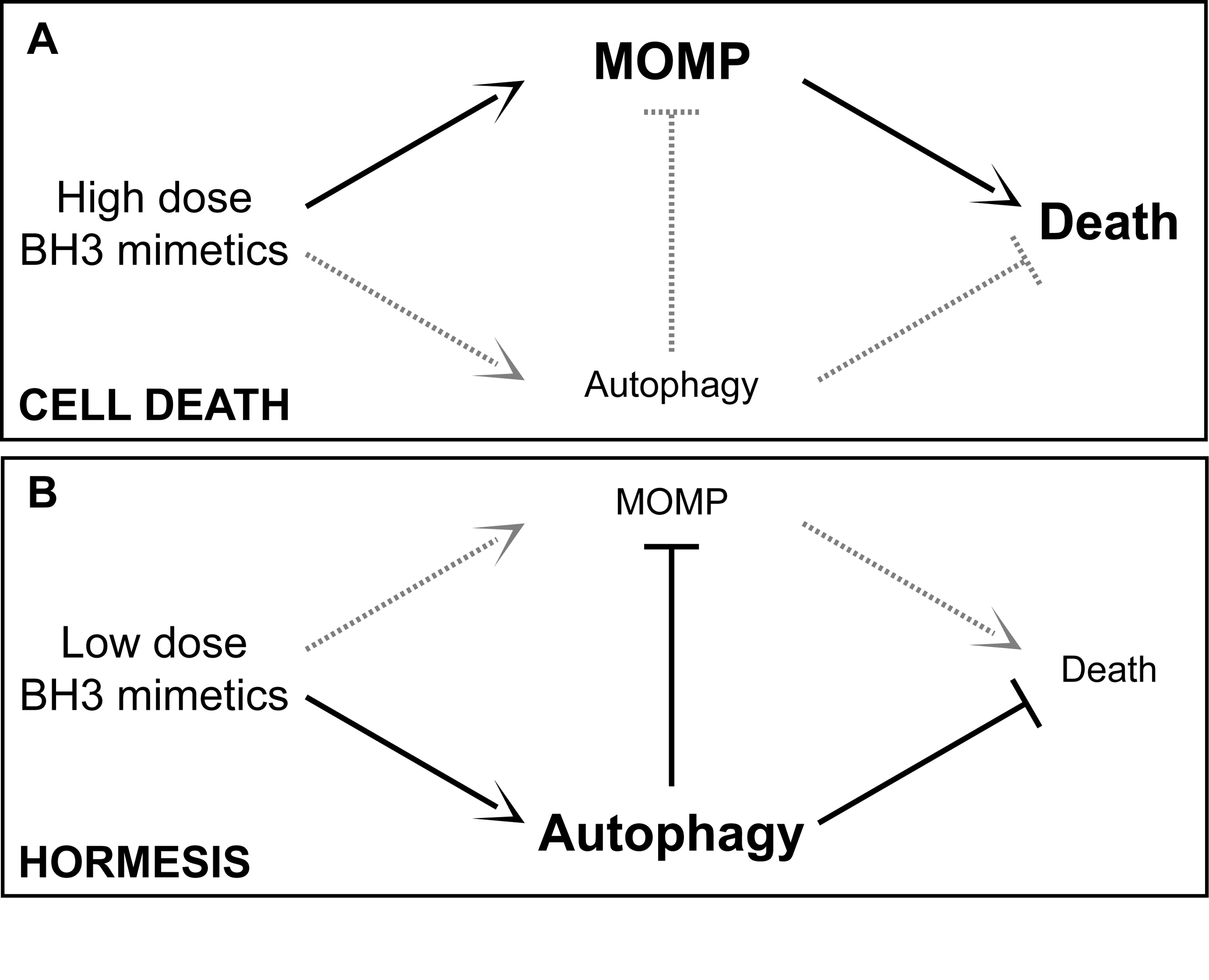
{kind=link}
11. Anti-aging effects of Autophagy on Proliferating Cells
– Autophagy has cytoprotective effects and other unique effects in dividing cells:
- Cytoprotective effects – see #10 above
- Reduced stem cell attrition
- Reduced ROS-induced cellular senescence
- Reduced oncogenic transformation
- Improved genetic stability
- Increased p62 degradation
- Anti-cancer effects via increased oncogene-induced senescence and oncogene-induced apoptosis
With aging, there is a decline in bone marrow stem cell function (hematopoeitic stem cells and mesenchymal stem cells) and stem cell number (MSCs only). Rapamycin restores the self-renewal capability of hematopoietic stem cells (HSCs). This improves the function of the immune system, of course assuming a lower dose of rapamycin than the immunosuppressive rapamycin dose given for preventing organ transplant rejection. Rapamycin can also reverse the stem cell loss that occurs in hair follicles and thereby prevent alopecia. mTOR accelerates cellular senescence by increasing the expression of p16/INK4a, p19/Arf, and p21/Cip1. These are all markers of cellular senescence and up regulating these tumor suppressors induces cellular senescence.
The tumor suppressor PTEN is just the opposite, however. Loss of the tumor suppressor PTEN induces a unique type of cellular senescence called “PTEN loss-induced cellular senescence” (PICS). PICS occurs with mTOR activation and can be reduced by inhibiting MDM2, which leads to an increase in p53 expression. This would inhibit autophagy. Rapamycin can preclude permanent (irreversible) cell-cycle arrrest due to inducible p21 expression. In this aspect, mTOR decreases proliferative potential and mediates stem cell attrition via senescence. Rapamycin can suppress this. This effect may be mediated by autophagy or by an autophagy-independent effect of mTOR inhibition.
More importantly, several oncogenes suppress autophagy. This includes Akt1, PI3K, Bcl-2 family anti-apoptotic proteins. Most of the proteins that stimulate autophagy also inhibit oncogenesis. This includes DAPK1, PTEN, TSC1, TSC2, LKB1/STK11, and Beclin-1. Autophagy can suppress oncogenesis through cell-autonomous effects described below:
- Improved quality control of mitochondria (less baseline ROS production)
- Enhanced genetic stability
- Removal of potentially oncogenic protein p62 via autophagy.
- Autophagy up regulation results in oncogene-induced senescence (via Ras)
The diagram below shows the beneficial effects of autophagy on all cell types, specific benefits in proliferating cells, and specific benefits in post-mitotic cells.
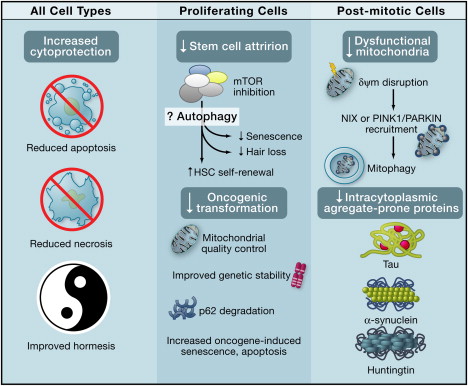
Systemic Anti-Aging Effects of Autophagy Image source
12. Autophagy can reduce age-related dysfunction through systemic effects –
Autophagy also confers several beneficial anti-aging effects that are not due to cytoprotection, or other localized effects within the cell itself. This includes the following systemic benefits of autophagy:
- Defense against infections
- Innate immunity
- Inhibition of pro-inflammatory signaling
- Neuroendocrine effects of autophagy
Autophagy in dying antigen-presenting cells improves the presentation of the antigens to dendritic cells. In dendritic cells, autophagy improves antigen presentation to T cells. Autophagy in dying cells is also required for macrophage clearance of these dead/dying cells. This is how autophagy reduces inflammation. Autophagy helps keep ATP production going in these dying cells, providing energy for the key step in the lysophosphatidylcholine “find me” signaling as well as the phosphatidylserine “flip flop” that is the “eat me” recognition signal for macrophage ingestion of the dying/dead cells. By helping macrophages find these cells and recognize that they are ready for macrophage ingestion, these cells do not rupture and spill their intracytoplasmic contents (this is what causes the inflammation with necrosis, where cell membrane rupture occurs).
When autophagy is working hand-in-hand with apoptosis, no inflammation occurs when a cell dies. This is a key beneficial role of autophagy in reducing inflammation. The decline in autophagy seen in aging may be in part the cause of age-induced type-2 diabetes. Here the peripheral tissues become insulin resistant. This may be due to the hepatic suppression of the Atg7 gene, which results in ER stress and insulin resistance. Induction of autophagy in specific neural populations may be sufficiency to reduce pathological aging.
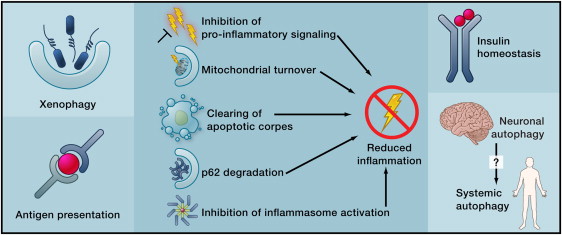
More effects of autophagy Image source
Beyond its cell-autonomous action, autophagy can reduce age-related dysfunctions through systemic effects. Autophagy may contribute to the clearance of intracellular pathogens and the function of antigen-presenting cells (left), reduce inflammation by several mechanisms (middle), or improve the function of neuroendocrine circuits (right).
13. Autophagy is necessary for maintaining the health of pools of adult stem cells
Frequent readers of this blog know that the writers believe that age-related decline of the health and differentiation capability of adult stem cells and increasing sensescence of those cells may be responsible for many of the effects we associate with aging. Thus, the positive roles of autophagy in keeping stem cells viable is of great interest to us.
See the comments under 11 above. Also, the June 2013 review publication Autophagy in stem cells provides “a comprehensive review of the current understanding of the mechanisms and regulation of autophagy in embryonic stem cells, several tissue stem cells (particularly hematopoietic stem cells), as well as a number of cancer stem cells.” Another such review is the June 2012 e-publication Tightrope act: autophagy in stem cell renewal, differentiation, proliferation, and aging.
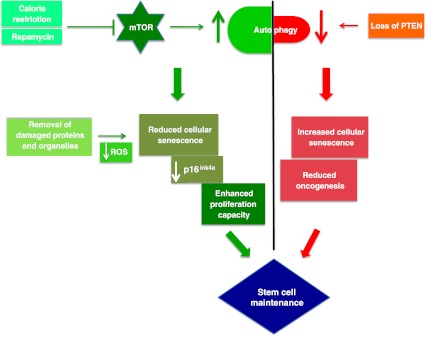
Image Source “Tightrope act inhibition of mTOR via caloric restriction (CR) or rapamycin induces autophagy. Autophagy clears away damaged proteins and organelles like defective mitochondria, thereby decreasing ROS levels and reducing genomic damage and cellular senescence, thus playing a crucial role in enhancing stem cell longevity. CR may also have a role in maintaining low levels of p16ink4a, a tumor suppressor protein, thus reducing the risk of cancer and promoting proliferation of stem cells. Oncogenesis is countered by loss of PTEN which elicits a p53-dependent prosenescence response to decrease tumorigenesis(ref)”
Only now are studies beginning to emerge that characterize the detailed roles of autophagy in maintaining stem cell health and differentiation viability. Autophagy in stem cells recapitulates the current state of understanding: “As a major intracellular degradation and recycling pathway, autophagy is crucial for maintaining cellular homeostasis as well as remodeling during normal development, and dysfunctions in autophagy have been associated with a variety of pathologies including cancer, inflammatory bowel disease and neurodegenerative disease. Stem cells are unique in their ability to self-renew and differentiate into various cells in the body, which are important in development, tissue renewal and a range of disease processes. Therefore, it is predicted that autophagy would be crucial for the quality control mechanisms and maintenance of cellular homeostasis in various stem cells given their relatively long life in the organisms. In contrast to the extensive body of knowledge available for somatic cells, the role of autophagy in the maintenance and function of stem cells is only beginning to be revealed as a result of recent studies. Here we provide a comprehensive review of the current understanding of the mechanisms and regulation of autophagy in embryonic stem cells, several tissue stem cells (particularly hematopoietic stem cells), as well as a number of cancer stem cells. We discuss how recent studies of different knockout mice models have defined the roles of various autophagy genes and related pathways in the regulation of the maintenance, expansion and differentiation of various stem cells. We also highlight the many unanswered questions that will help to drive further research at the intersection of autophagy and stem cell biology in the near future.”
Another very-recent finding related to autophagy and stem cells is reported in the March 31, 2013 paper FIP200 is required for maintenance and differentiation of postnatal neural stem cells. “These data reveal that FIP200-mediated autophagy contributes to the maintenance and functions of NSCs through regulation of oxidative state.” FIP200 is “a gene essential for autophagy induction in mammalian cells.”
Exercising control over autophagy may prove useful for efficiently generating induced pluripotent stem cells. According to the 2012 publication Autophagy in stem cell maintenance and differentiation: “We also discuss a possible role for autophagy during cellular reprogramming and induced pluripotent stem (iPS) cell generation by taking advantage of ATP generation for chromatin remodeling enzyme activity and mitophagy. Finally, the significance of autophagy modulation is discussed in terms of augmenting efficiency of iPS cell generation and differentiation processes.”
A steady stream of research continues to reveal new insights on the roles that autophagy plays in stem cells. For example, the April 2013 publication FOXO3A directs a protective autophagy program in haematopoietic stem cells reports: “Here we identify autophagy as an essential mechanism protecting HSCs from metabolic stress. We show that mouse HSCs, in contrast to their short-lived myeloid progeny, robustly induce autophagy after ex vivo cytokine withdrawal and in vivo calorie restriction. We demonstrate that FOXO3A is critical to maintain a gene expression program that poises HSCs for rapid induction of autophagy upon starvation. Notably, we find that old HSCs retain an intact FOXO3A-driven pro-autophagy gene program, and that ongoing autophagy is needed to mitigate an energy crisis and allow their survival. Our results demonstrate that autophagy is essential for the life-long maintenance of the HSC compartment and for supporting an old, failing blood system.”
14. Autophagy is a key step in activating the Nrf2 pathway. And Nrf2 expression can in turn regulate autophagy.
The importance of the Nrf2 stress-response pathway and its role in generating health has been one of the frequent topics of discussion in this blog. See specifically the blog entries The pivotal role of Nrf2. Part 1, Part 2, Part 3, and Nrf2 and cancer chemoprevention by phytochemicals. We know now that autophagy plays a key role in Nrf2 activation, via p62-dependent autophagic degradation of Keap1. See, for example, the 2012 publication Sestrins Activate Nrf2 by Promoting p62-Dependent Autophagic Degradation of Keap1 and Prevent Oxidative Liver Damage. We also know that, in turn, Nrf2 expression can regulate autophagy. See for example the March 2013 publication Regulation of Cigarette Smoke (CS)-Induced Autophagy by Nrf2.
15. Autophagy and aging
We are starting to understand why autophagy stops working well when a person grows old – why autophagy does not work as well as you age. Among the reasons are:
a. Failure to form autophagosomes – with aging, there appears to be a failure for autophagosomes to form, possibly due to macroautophagy enhancers (glucagon).
b. Failure of fusion – with aging, there appears to be a failure of lysosomes to fuse with autophagosomes.
c. Negative signaling from insulin or insulin receptors – with aging, insulin signaling or insulin receptor signaling activates mTOR in cells.
d. Mitophagy does not work as well in aging.
e. Autophagy decline probably also results in energy (ATP production) decline.
16. Practical interventions to promote autophagy
There are a number of practical ways to promote autophagy. Specifically, in partial recap of the above:
- Fasting activates Autophagy – caloric restriction affects 5 molecular pathways that activate autophagy
- Sunlight, Vitamin D and Klotho activate Autophagy – there are three ways through which UV light, Vitamin D, and the Klotho pathway activate autophagy via inhibiting the insulin/IGF-1 pathway
- Rapamycin activates Autophagy – there are two ways through which mTOR inhibitors activate autophagy – TORC1 and TORC2 mechanisms
- Caffeine activates Autophagy – Caffeine can activate autophagy via an mTOR-dependent mechanism
- Green tea activates Autophagy – ECGC can activate autophagy via an mTOR-dependent mechanism
- Metformin activates Autophagy – metformin can activate autophagy via AMPK activation – mTOR-dependent and mTOR-independent mechanisms
- Lithium activates Autophagy – lithium and other compounds can activate autophagy by inhibiting inositol monophosphate and lower IP3 levels – an mTOR-independent mechanism
- Resveratrol activates Autophagy – there are four 4 ways through which resveratrol can activate autophagy – via mTOR-dependent and mTOR-independent mechanisms
- Spermidine activates Autophagy – how spermidine activates autophagy via histone protein deacetylation – mTOR-indepdendent mechanism
- Hypoxia activates Autophagy – intermittent hypoxia can increase autophagy via HIF-1a
- Phytosubstances which activate the Nrf2 pathway can activate Autophagy. These are many and include soy products and hot chili peppers.
In addition, these lesser-known substances can also activate autophagy:
Amiodarone low dose Cytoplasm – midstream yes Calcium channel blocker => TORC1 inhibition. Also, a mTOR-independent autophagy inducer
- Fluspirilene low dose Cytoplasm – midstream yes Dopamine antagnoists => mTOR-dependent autophagy induction
- Penitrem A low dose Cytoplasm – midstream yes high conductance Ca++activated K+ channel inhibitor => mTOR-dependent autophagy inducer
- Perihexilenelowdose Cytoplasm- midstream yes 1. TORC1 inhibition
- Niclosamidelowdose Cytoplasm- midstream yes 1. TORC1 inhibition
- Trehalose 100 mM Cytoplasm – midstream supplement 1. activates autophagy via an mTOR-independent mechanism
- Torin-1 low dose Cytoplasm – midstream no 1. mTOR inhibition (much more potent than rapamycin)
- Trifluoperazine low dose Cytoplasm – midstream yes Dopamine antagonists => mTOR-dependent autophagy induction
Wrapping it up
Here are some of the main points above covered:
- Autophagy is like having a Pac man inside each of your cells, chasing down, eating up and recycling dysfunctional organelles, proteins and protein aggregates. It has three forms: i. chaperone-mediated autophagy, ii. microautophagy and iii. macroautophagy. The last is the most important one.
- Autophagy is a stress response and behaves according to the principles of hormesis.
- Autophagy can retire and eat up old mitochondria which have become electron-leaking engines.
- Autophagy solves the problem of high baseline levels of reactive oxygen and nitrogen species.
- Autophagy does not require proteins to be unfolded for it to work and therefore can perform housekeeping tasks undoable by the other cell-level house cleaning system, the ubiquitin-proteasome system.
- Autophagy gets rid of the protein aggregates that can make you loose your memory or walk slow as you grow old – those associated with Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, ALS, CTE, and other neurodegenerative conditions.
- Autophagy keeps adult stem cells healthy and facilitates their capability to differentiate to make normal somatic body cells.
- Autophagy prevents inflammation – it works hand-in-hand with apoptosis to help the body get rid of dying cells without inducing cell rupture and inflammation.
- Autophagy prevents cancer – it helps maintain genetic stability, prevents epigenetic gene silencing. And it helps promote oncogene-induced cellular senescence for cancer prevention.
- Autophagy saves the lives of cells by preventing unnecessary cellular apoptosis and cell necrosis.
- Autophagy is involved in Nrf2 activation and to some extent Nrf2 expression negatively regulates autophagy.
- Autophagy keeps your bone marrow stem cell population alive and functional.
- Autophagy helps with infections – it helps clear intracellular pathogens such as bacteria and viruses.
- Autophagy improves the innate immune response.
- We are starting to understand why autophagy declines with aging.
- While autophagy declines with aging, it can exercise multiple effects to slow aging down. It inhibits the major mechanisms of aging such as cellular senescence, protein aggregate build-up, stem cell loss, epigenetic gene silencing, telomere shortening, and oxidative damage to proteins, lipids, and DNA.
- There are many practical ways to activate Autophagy like consuming green tea and caffeine, and some less-practical ones.
Source: Autophagy – the housekeeper in every cell that fights aging | AGINGSCIENCES™ – Anti-Aging Firewalls™